-
Europium-Doped Ba7F12Cl2, a Single Component Near-UV Excited Tunable White Phosphor
H. Hagemann, H. Bill, J.M. Rey, F. Kubel, L. Calame and D. Lovy
The Journal of Physical Chemistry C, 119 (1) (2015), p141-147


DOI:10.1021/jp510301p | unige:44953 | Abstract | Article HTML | Article PDF | Supporting Info

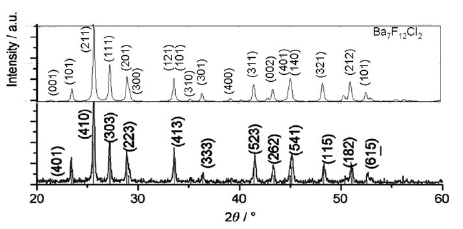
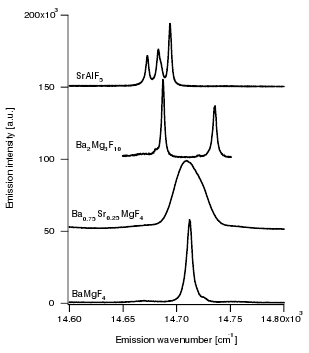
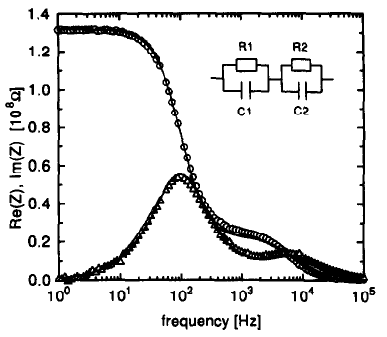
Europium doped crystalline Ba7F12Cl2 phosphors have been prepared at temperatures between 650 and 900 °C using alkali chloride fluxes, yielding both disordered (with the incorporation of small amounts of Na) and ordered crystal modifications. The white emission spectrum excited in the near UV consists roughly of two broad emission bands at ca 450 and 590 nm, as well as weak sharp Eu2+ 4f-4f emission bands around 360 nm. The incorporation of Eu2+ is further studied using EPR spectroscopy on single crystals, and reveals a significant zero field splitting. The emission spectrum can be significantly tuned by varying the excitation wavelength between 300 and 390 nm. Fine tuning may also be achieved by chemical substitutions to form Ba7-xMyF12Cl2-zBrz (M = Na, Ca,Eu). Quantitative measurements of the light produced using commercial near UV LEDs show that the color temperature ranges between 4000 and 9700 K with CIE chromaticity coordinates close to the ideal values of x=y=0.333. The best color rendering index (CRI) found was 0.83, and the highest light to light conversion yield was 171 lumen/W. These results show that the title compound is a very promising candidate for white light generation using near UV LED excitation.